# Packages
library(ggtext)
library(ggtree)
library(ape)
library(ggplot2)Durante el mes de abril se celebró el #30DayChartChallenge, un reto de visualización de datos organizado por @dr_xeo y @CedScherer en el que cada día se debía presentar un gráfico diferente siguiendo la temática de ese día. En el siguiente enlace se pueden ver todas las contribuciones realizadas en R a lo largo del challenge.
El día 16 de abril, bajo la temática de árboles, desde El arte del dato bien contado optamos por un árbol filogenético. Este tipo de gráficos se utilizan en Biología para representar las relaciones evolutivas de una o varias especies que parten de un ancestro común. Los árboles filogenéticos requieren un formato específico de almacenamiento de datos alejado de los tipos de objetos de R a los que estamos acostumbrados. En concreto, la clase phylo es la utilizada para el análisis de datos filogenéticos.
Librerías
El primer paso será cargar las librerías necesarias. ggtext, la utilizaremos para dar estilo al título y subtítulo de nuestro plot, por lo que no es imprescindible para el árbol en sí. Sin embargo, como veis parte del encanto de esta visualización en conjunto es la tipografía y su estilo, por lo que recomendamos esta librería 100%!
Por otro lado, ggtree, es el paquete principal que permite analizar y visualizar esta clase de árboles. Se trata de una librería desarrollada por Guangchuang Yu. En su libro, explica en detalle cómo manipular este tipo de datos así como todas las posibilidades que ofrece en cuanto a visualización y tipos de árboles.
Finalmente, la librería ape nos servirá para convertir el dataset principal en un objeto de clase filogenética.
Datos
A continuación cargamos los datos. Esta vez hemos elegido un dataset sencillo ya que gran parte del trabajo recaía sobre el árbol en sí (y tengamos en cuenta que el reto consistía en hacer un gráfico por día durante 30 días 😅). Simplemente, debemos calcular las distancias entre las distintas flores y transformar estas distancias en un árbol filogenético. La función bionj de la librería ape convierte objetos de tipodist en objetos de clase phylo por lo que es la opción que utilizaremos en nuestro caso.
Por último, la función groupOTU permite asignar ramas y nodos a diferentes grupos basados en grupos especificados por el usuario. En nuestro caso las especies de Iris: Setosa, Virgínica y Versicolor.
# Data
data(iris)
rn <- paste0(iris[,5], "_", 1:150)
rownames(iris) <- rn
d_iris <- dist(iris[,-5], method="man")
tree_iris <- ape::bionj(d_iris)
grp <- list(setosa = rn[1:50],
versicolor = rn[51:100],
virginica = rn[101:150])
tree_iris <- groupOTU(tree_iris, grp, "Species")Plot
El árbol se representa utilizando la librería ggtree y asignando la variable Species al color del árbol. Tanto el fondo como la escala de colores proviene de una paleta cualitativa de Carto Colors, caracterizada por sus tonos pastel. Si quieres saber más sobre cómo elegir colores en R, en el último post hablamos en detalle sobre ello!
# Plot
ggtree(tree_iris, aes(color=Species), layout = 'daylight', branch.length='none') +
scale_color_manual(values=c('#7bbcb0','#f8b58b','#e38191')) +
theme(legend.position = 'none',
panel.border= element_blank(),
panel.grid = element_line(color='#a33131', size=0.25),
plot.margin = margin(20, 100, 20, 100),
plot.background = element_rect(fill='#242629', color='#242629'),
panel.background = element_rect(fill='transparent', color='transparent'),
plot.title = element_markdown(size=10, color='white', family='mono',
hjust = 0, margin=margin(10,0,20,0)),
plot.subtitle = element_markdown(size=6, family='mono', color='#ffffff', margin=margin(0,0,30,0)),
plot.caption = element_text(size=7.5, hjust=0.5, color='#ffffff', family='mono', margin=margin(30,0,10,0))) +
labs(title='Phylogenetic Tree of Iris Species. <span style="color: #7bbcb0">Setosa.</span>
<span style="color: #f8b58b">Versicolor.</span>
<span style="color: #e38191">Virginica.</span><br>',
caption = "Paula L. Casado (@elartedeldato) | Data: R datasets \n#30DayChartChallenge | Day 16: Tree")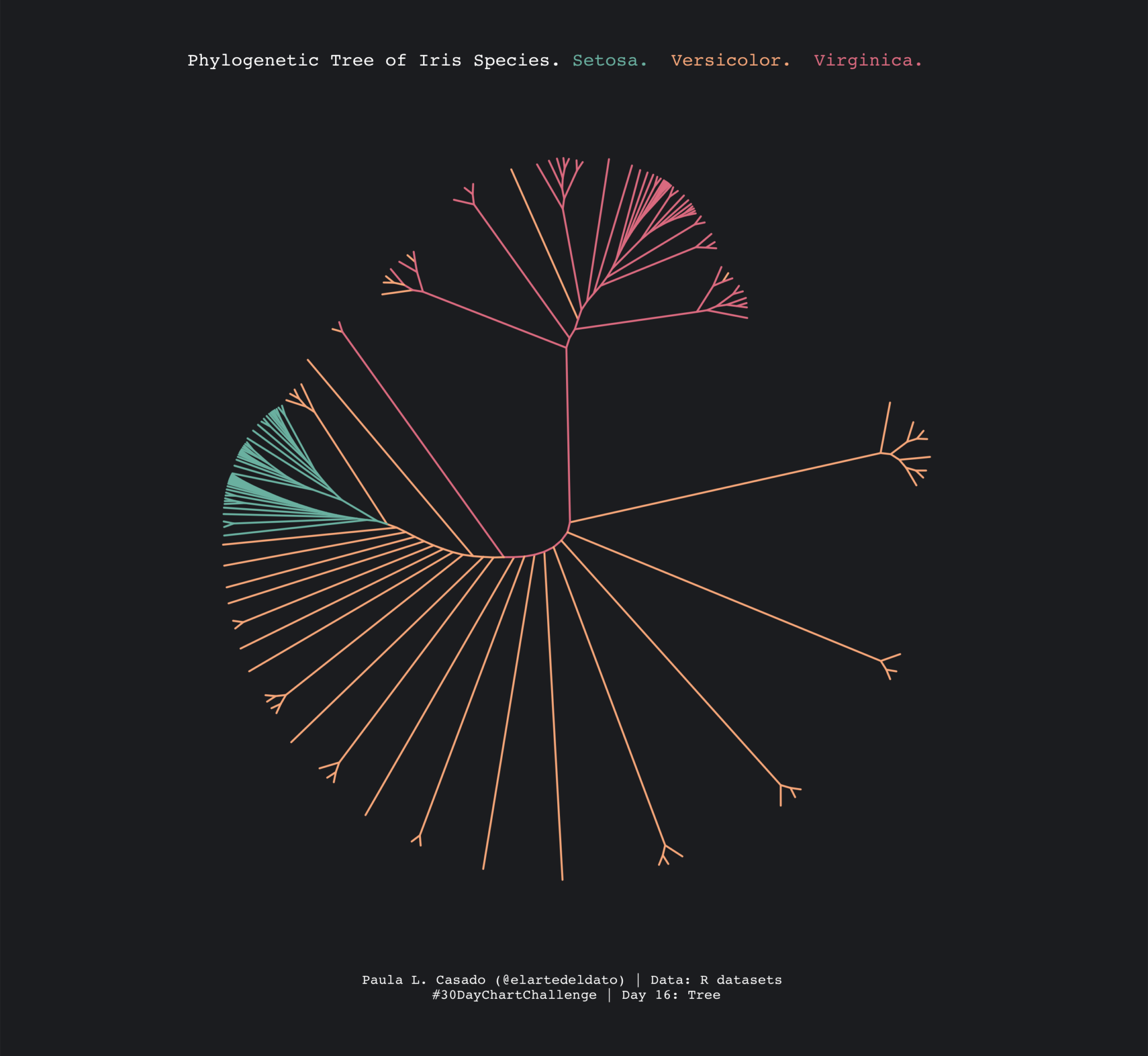
El resultado final también depende en gran medida del tamaño del gráfico, la tipografía, la posición de los elementos y el grosor de líneas. Todo ello, no es sencillo de escoger, por eso recomendamos dedicarle el tiempo necesario a los últimos detalles, que pueden ser la clave del éxito de nuestra visualización. 🤗
Bibliografía: https://yulab-smu.top/treedata-book/index.html
Contenido relacionado


